The Use Of RNA-seq For The Study Of Physiological Adaptations Of Halophiles In Extreme Environments For Astrobiological Data Interpretation
The purpose of this paper aims to discuss the utility of RNA-seq, a transcriptomic analysis tool, to study the details of halophile adaptations within terrestrial evolution and for projected Martian ecological stresses and applications for transcriptomics for astrobiology research.
The strategies shown here involve study of modern-day cells, which evolved from ancient cells. The modernday cell would be subjected to prebiotic world stress conditions or a simulated Martian environment. RNA-seq analysis will be performed on the stressed cell, generating a list of differentially expressed genes.
If commonalities in gene expression existed between extant and extinct cells, these genes might have also been upregulated in extinct cells on Earth or, hypothetically, in extinct cells on Mars (if they existed). The gene expression data will help us to hypothesize whether the extinct cells on Earth or Mars (if they existed) adapted to similar types of stresses.
Environments This paper discusses the potential of next-generation sequencing tools like RNA-seq to study gene expression in halophiles for astrobiology research. RNA-seq is used to quantify mRNA in a given sample at a particular time point.
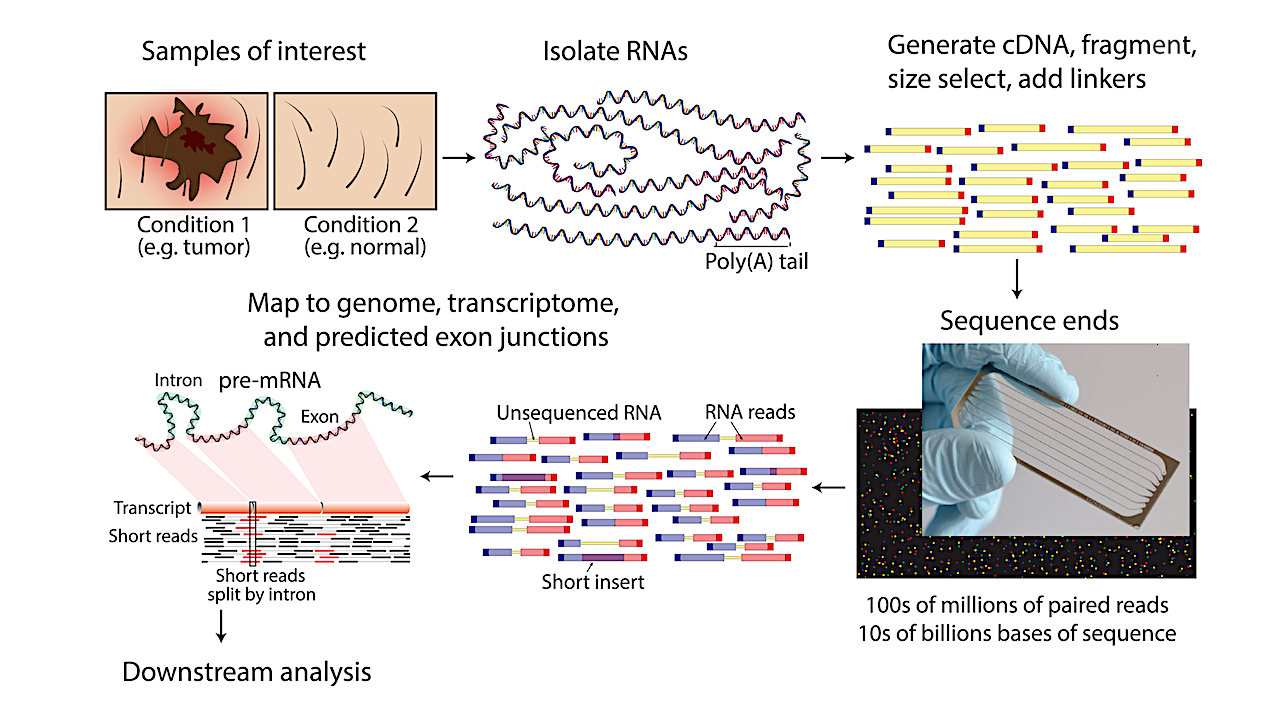
In this technique, mRNA is extracted from ‘control’ and ‘treated’ samples. Then, the mRNAs are converted to cDNAs (complementary DNAs or ‘photocopies of the genes’) using the enzyme reverse transcriptase. After that, cDNAs from various samples are ‘tagged’ with short DNA sequences (barcodes) to distinguish among various samples.
After that, the barcoded cDNAs are sequenced using a high-throughput DNA sequencer (like Illumina miSeq ™ or Illumina hiSeq ™ ). It is possible to obtain between 10 million and 100 million sequences per sample within 24 hours or less. The sequenced cDNAs could be searched against public databases like GenBank to catalog a list of differentially upregulated or downregulated genes amongst various treatments or samples.
We propose that RNA-seq data can unpack the gene expressions of halophilic microorganisms to show the links between the ecological settings of this planetary analog site and what gene expressions need to be activated in life elsewhere to survive. RNA-seq is a powerful tool for studying microbial diversity without the need to conduct additional sequencing (Cox et al., 2017).
RNA-seq can also be used to study microbes whose genome is not sequenced or when no reference genome is available (Cox et al., 2017); this is especially important in astrobiology research where the collected species samples might not have any background information available. Because of its ease of use, RNA-seq has already replaced microarray in transcriptomic studies and has become the most potent tool in bacterial gene regulation studies (Poulsen and Vinther, 2018).
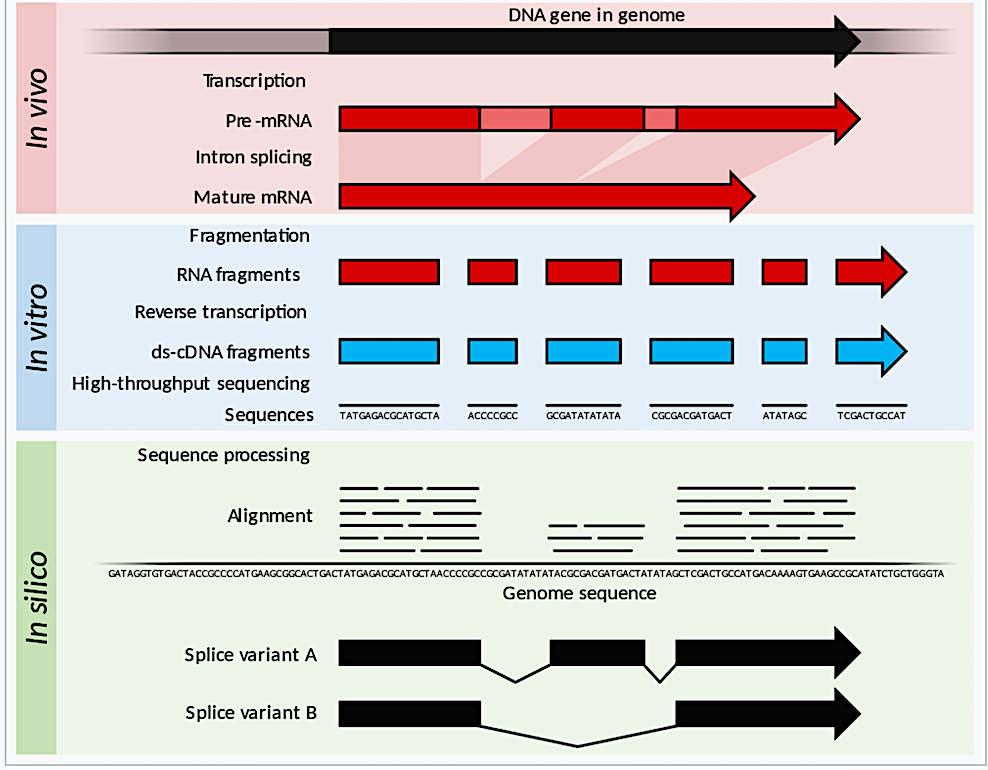
Summary of RNA-Seq. Within the organism, genes are transcribed and (in an eukaryotic organism) spliced to produce mature mRNA transcripts (red). The mRNA is extracted from the organism, fragmented and copied into stable ds-cDNA (blue). The ds-cDNA is sequenced using high-throughput, short-read sequencing methods. These sequences can then be aligned to a reference genome sequence to reconstruct which genome regions were being transcribed. This data can be used to annotate where expressed genes are, their relative expression levels, and any alternative splice variants. – Wikipedia
The use of RNA-seq for the study of Physiological Adaptations of Halophiles in Extreme Environments for Astrobiological Data Interpretation, Frontiers in Astronomy and Space Sciences
Astrobiology, genomics