Integrating Phylogenies Into Single-cell RNA Sequencing Analysis Allows Comparisons Across Species, Genes, And Cells
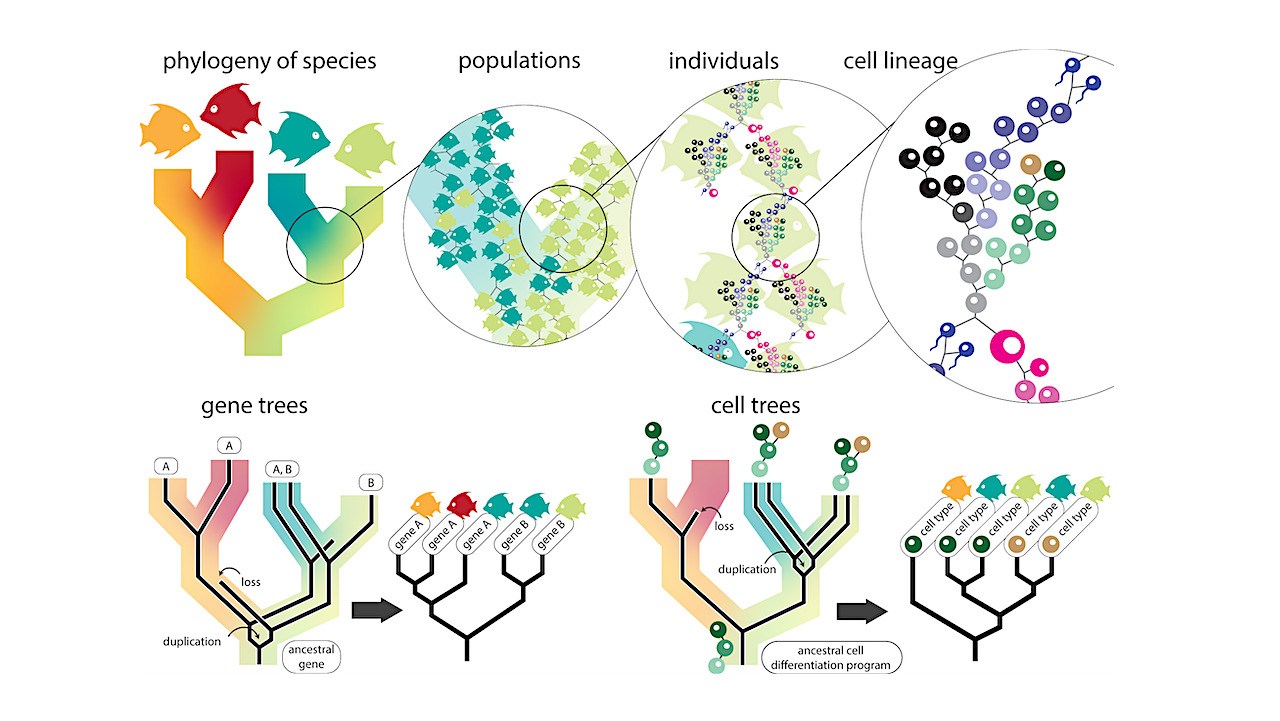
Abstract: Comparisons of single-cell RNA sequencing (scRNA-seq) data across species can reveal links between cellular gene expression and the evolution of cell functions, features, and phenotypes. These comparisons evoke evolutionary histories, as depicted by phylogenetic trees, that define relationships between species, genes, and cells.
This Essay considers each of these in turn, laying out challenges and solutions derived from a phylogenetic comparative approach and relating these solutions to previously proposed methods for the pairwise alignment of cellular dimensional maps.
This Essay contends that species trees, gene trees, cell phylogenies, and cell lineages can all be reconciled as descriptions of the same concept—the tree of cellular life. By integrating phylogenetic approaches into scRNA-seq analyses, challenges for building informed comparisons across species can be overcome, and hypotheses about gene and cell evolution can be robustly tested.
Introduction (excerpt)
Single-cell RNA sequencing (scRNA-seq) generates high-dimensional gene expression data from thousands of cells from an organ, tissue, or body [1]. Single-cell expression data are increasingly common, with new animal cell atlases being released every year [2–6]. The next steps will be to compare such atlases across species [2], identifying the dimensions in which these results differ and associating these differences with other features of interest [7]. Because all cross-species comparisons are inherently evolutionary comparisons, such analyses present an opportunity to integrate approaches from the field of evolutionary biology, and especially phylogenetic biology [8].
Drawing concepts, models, and methods from these fields will help to overcome central challenges with comparative scRNA-seq analysis, especially in how to draw coherent comparisons over thousands of genes and cells across species. In addition, this synthesis of concepts will help avoid the unnecessary reinvention of analytical methods that have already been rigorously tested in evolutionary biology for other types of data, such as morphological and molecular data.
Comparative gene expression analysis has been used for decades to answer evolutionary questions such as how changes in gene expression are associated with the evolution of novel functions and phenotypes [9]. The introduction of scRNA-seq technology has led to a massive increase in the scale of these experiments [1], from working with a few genes or a few tissues, to assays that cover the entire transcriptome, across thousands of cells in a dissociation experiment.
Comparative scRNA-seq analysis therefore enables evolutionary questions to be scaled up, for example: how has the genetic basis of differentiation evolved across cell populations and over time; what kinds of cells and gene expression patterns were likely present in the most recent common ancestor; what changes in cell transcriptomes are associated with the evolution of new ecologies, life-histories, or other features; how much variation in cellular gene expression do we observe over evolutionary time; which changes in gene expression are significant (i.e., larger or smaller than we expect by chance); which genes show patterns of correlated expression evolution; and can evolutionary screens detect novel interactions between genes?

Types of scRNA-seq data that can be mapped onto a phylogeny. Several types of scRNA-seq data could potentially be mapped onto a phylogeny. Nine types of data are shown, along with example questions that can be addressed and caveats to be considered. PLoS Biology
Full paper Integrating phylogenies into single-cell RNA sequencing analysis allows comparisons across species, genes, and cells, PLoS Biology (open access)
Astrobiology